model_atac
Yunqi Yang
8/11/2020
Last updated: 2020-08-18
Checks: 6 1
Knit directory: gene_level_fine_mapping/
This reproducible R Markdown analysis was created with workflowr (version 1.6.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200622) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Using absolute paths to the files within your workflowr project makes it difficult for you and others to run your code on a different machine. Change the absolute path(s) below to the suggested relative path(s) to make your code more reproducible.
| absolute | relative |
|---|---|
| /Users/nicholeyang/Desktop/gene_level_fine_mapping/data/train_all.RData | data/train_all.RData |
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 038b52c. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.DS_Store
Ignored: analysis/.RData
Ignored: analysis/.Rhistory
Untracked files:
Untracked: analysis/atac_eqtl.Rmd
Untracked: data/hic_eqtl.RData
Untracked: data/train_add_hic.RData
Untracked: data/train_all.RData
Unstaged changes:
Modified: analysis/add_hic_feature.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/model_atac.Rmd) and HTML (docs/model_atac.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 038b52c | yunqiyang0215 | 2020-08-18 | wflow_publish(“analysis/model_atac.Rmd”) |
| html | a32fa5b | yunqiyang0215 | 2020-08-11 | Build site. |
| Rmd | 0e0f656 | yunqiyang0215 | 2020-08-11 | wflow_publish(“analysis/model_atac.Rmd”) |
load('/Users/nicholeyang/Desktop/gene_level_fine_mapping/data/train_all.RData')head(train_all_sig) gene_name snp_loc38 variant_id UTR5 UTR3 exon intron
1 A1BG chr19:57866502 chr19_57866502_T_C_b38 0 0 0 0
2 A1BG chr19:58059544 chr19_58059544_T_G_b38 0 0 0 0
3 A1BG chr19:58170494 chr19_58170494_G_T_b38 0 0 0 0
4 A1BG chr19:58228973 chr19_58228973_T_G_b38 0 0 0 0
5 A1BG chr19:58330182 chr19_58330182_C_T_b38 0 0 0 0
6 A1BG chr19:58359927 chr19_58359927_G_A_b38 0 0 0 0
upstream tss_dist_to_snp y Mon Mac0 Mac1 Mac2 Neu MK EP Ery FoeT nCD4 tCD4
1 0 481132 0 0 0 0 0 0 0 0 0 0 0 0
2 0 288090 0 0 0 0 0 0 0 0 0 0 0 0
3 0 177140 0 0 0 0 0 0 0 0 0 0 0 0
4 0 118661 0 0 0 0 0 0 0 0 0 0 0 0
5 0 17452 1 0 0 0 0 0 0 0 0 0 0 0
6 0 6428 1 0 0 0 0 0 0 0 0 0 0 0
aCD4 naCD4 nCD8 tCD8 nB tB correlation
1 0 0 0 0 0 0 0
2 0 0 0 0 0 0 0
3 0 0 0 0 0 0 0
4 0 0 0 0 0 0 0
5 0 0 0 0 0 0 0
6 0 0 0 0 0 0 0correlation by distance
Comment: ATAC features are mostly concentrated in the range < 5e5.
par(mfrow = c(1,2))
plot(train_all_sig$tss_dist_to_snp, train_all_sig$correlation, xlab = 'distance', ylab = 'correlation')
plot(train_all_sig[train_all_sig$correlation>0.4, ]$tss_dist_to_snp, train_all_sig[train_all_sig$correlation>0.4, ]$correlation, xlab = 'distance', ylab = 'correlation')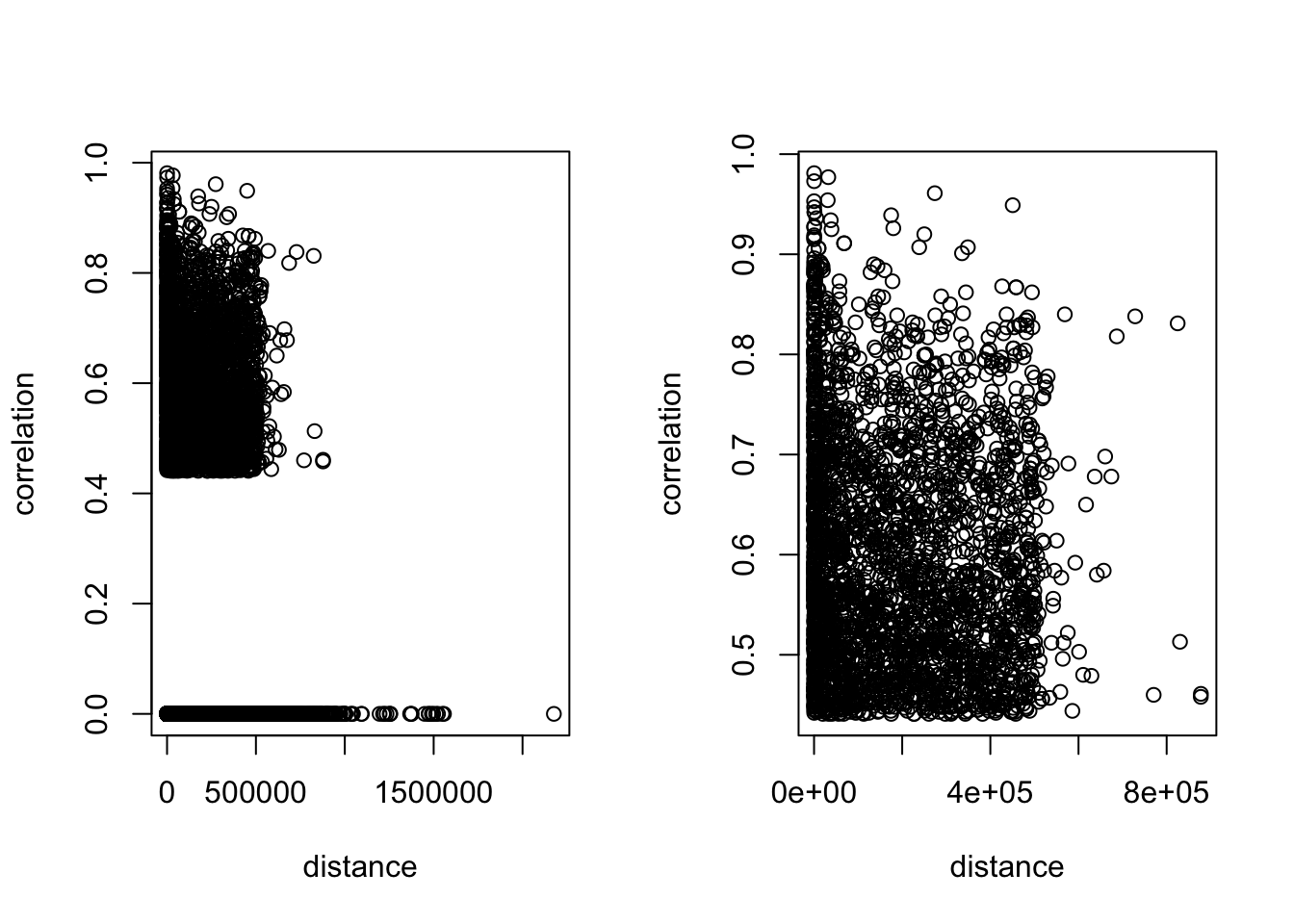
direct regress correlation on y
Complete separation?
https://courses.ms.ut.ee/MTMS.01.011/2018_spring/uploads/Main/GLM_slides_7_binary_response_ii.pdf
fit1 = glm(y ~ correlation, data = train_all_sig, family = "binomial")
fit2 = glm(y ~ correlation + tss_dist_to_snp, data = train_all_sig, family = "binomial")Warning: glm.fit: fitted probabilities numerically 0 or 1 occurredsummary(fit1)
Call:
glm(formula = y ~ correlation, family = "binomial", data = train_all_sig)
Deviance Residuals:
Min 1Q Median 3Q Max
-0.8550 -0.3292 -0.3292 -0.3292 2.4258
Coefficients:
Estimate Std. Error z value Pr(>|z|)
(Intercept) -2.88817 0.01770 -163.16 <2e-16 ***
correlation 2.11891 0.08332 25.43 <2e-16 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
(Dispersion parameter for binomial family taken to be 1)
Null deviance: 29356 on 66456 degrees of freedom
Residual deviance: 28852 on 66455 degrees of freedom
AIC: 28856
Number of Fisher Scoring iterations: 5summary(fit2)
Call:
glm(formula = y ~ correlation + tss_dist_to_snp, family = "binomial",
data = train_all_sig)
Deviance Residuals:
Min 1Q Median 3Q Max
-1.6224 -0.1247 -0.0086 -0.0005 8.4904
Coefficients:
Estimate Std. Error z value Pr(>|z|)
(Intercept) -8.830e-02 2.822e-02 -3.129 0.00175 **
correlation 1.196e+00 1.100e-01 10.874 < 2e-16 ***
tss_dist_to_snp -4.016e-05 7.274e-07 -55.218 < 2e-16 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
(Dispersion parameter for binomial family taken to be 1)
Null deviance: 29356 on 66456 degrees of freedom
Residual deviance: 15847 on 66454 degrees of freedom
AIC: 15853
Number of Fisher Scoring iterations: 10#plot(fit1$fitted.values)
#plot(fit2$fitted.values)Try different threshold and make it binary
threshold = c(0.4, 0.5, 0.6, 0.7, 0.8, 0.9)
prop_pos = rep(NA, length(threshold))
prop_neg = rep(NA, length(threshold))
for (i in 1:length(threshold)){
train_all_sig$atac = ifelse(train_all_sig$correlation > threshold[i], 1, 0)
dat_pos = train_all_sig[train_all_sig$y == 1, ]
dat_neg = train_all_sig[train_all_sig$y == 0, ]
prop_pos[i] = sum(dat_pos$atac == 1)/dim(dat_pos)[1]
prop_neg[i] = sum(dat_neg$atac == 1)/dim(dat_neg)[1]
fit = glm(y ~ atac, data = train_all_sig, family = "binomial")
print(summary(fit))
}
Call:
glm(formula = y ~ atac, family = "binomial", data = train_all_sig)
Deviance Residuals:
Min 1Q Median 3Q Max
-0.5952 -0.3302 -0.3302 -0.3302 2.4233
Coefficients:
Estimate Std. Error z value Pr(>|z|)
(Intercept) -2.88167 0.01769 -162.89 <2e-16 ***
atac 1.24062 0.05357 23.16 <2e-16 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
(Dispersion parameter for binomial family taken to be 1)
Null deviance: 29356 on 66456 degrees of freedom
Residual deviance: 28931 on 66455 degrees of freedom
AIC: 28935
Number of Fisher Scoring iterations: 5
Call:
glm(formula = y ~ atac, family = "binomial", data = train_all_sig)
Deviance Residuals:
Min 1Q Median 3Q Max
-0.6360 -0.3315 -0.3315 -0.3315 2.4202
Coefficients:
Estimate Std. Error z value Pr(>|z|)
(Intercept) -2.87366 0.01754 -163.86 <2e-16 ***
atac 1.37837 0.05764 23.91 <2e-16 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
(Dispersion parameter for binomial family taken to be 1)
Null deviance: 29356 on 66456 degrees of freedom
Residual deviance: 28914 on 66455 degrees of freedom
AIC: 28918
Number of Fisher Scoring iterations: 5
Call:
glm(formula = y ~ atac, family = "binomial", data = train_all_sig)
Deviance Residuals:
Min 1Q Median 3Q Max
-0.7006 -0.3351 -0.3351 -0.3351 2.4116
Coefficients:
Estimate Std. Error z value Pr(>|z|)
(Intercept) -2.85166 0.01724 -165.4 <2e-16 ***
atac 1.57210 0.06927 22.7 <2e-16 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
(Dispersion parameter for binomial family taken to be 1)
Null deviance: 29356 on 66456 degrees of freedom
Residual deviance: 28970 on 66455 degrees of freedom
AIC: 28974
Number of Fisher Scoring iterations: 5
Call:
glm(formula = y ~ atac, family = "binomial", data = train_all_sig)
Deviance Residuals:
Min 1Q Median 3Q Max
-0.7971 -0.3387 -0.3387 -0.3387 2.4030
Coefficients:
Estimate Std. Error z value Pr(>|z|)
(Intercept) -2.82973 0.01699 -166.56 <2e-16 ***
atac 1.84600 0.09098 20.29 <2e-16 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
(Dispersion parameter for binomial family taken to be 1)
Null deviance: 29356 on 66456 degrees of freedom
Residual deviance: 29055 on 66455 degrees of freedom
AIC: 29059
Number of Fisher Scoring iterations: 5
Call:
glm(formula = y ~ atac, family = "binomial", data = train_all_sig)
Deviance Residuals:
Min 1Q Median 3Q Max
-0.9892 -0.3420 -0.3420 -0.3420 2.3950
Coefficients:
Estimate Std. Error z value Pr(>|z|)
(Intercept) -2.80959 0.01678 -167.41 <2e-16 ***
atac 2.34937 0.14651 16.04 <2e-16 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
(Dispersion parameter for binomial family taken to be 1)
Null deviance: 29356 on 66456 degrees of freedom
Residual deviance: 29166 on 66455 degrees of freedom
AIC: 29170
Number of Fisher Scoring iterations: 5
Call:
glm(formula = y ~ atac, family = "binomial", data = train_all_sig)
Deviance Residuals:
Min 1Q Median 3Q Max
-1.1483 -0.3445 -0.3445 -0.3445 2.3892
Coefficients:
Estimate Std. Error z value Pr(>|z|)
(Intercept) -2.79470 0.01665 -167.836 < 2e-16 ***
atac 2.72571 0.37198 7.327 2.35e-13 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
(Dispersion parameter for binomial family taken to be 1)
Null deviance: 29356 on 66456 degrees of freedom
Residual deviance: 29314 on 66455 degrees of freedom
AIC: 29318
Number of Fisher Scoring iterations: 5plot the proprotion of atac features using different threshold
Comment: shouldn’t use a too stringent threshold. Maybe 0.5-0.8.
prop_pos[1] 0.121582921 0.105701640 0.073939078 0.044780005 0.020046863 0.003644884prop_neg[1] 0.0384885652 0.0289223202 0.0163057366 0.0073463651 0.0019483838
[6] 0.0002395554prop_pos/prop_neg[1] 3.158936 3.654674 4.534544 6.095532 10.288970 15.215204prop_pos * dim(dat_pos)[1][1] 467 406 284 172 77 14prop_neg * dim(dat_neg)[1][1] 2410 1811 1021 460 122 15plot(threshold, prop_pos, type = 'b', ylab = 'proportion')
points(threshold, prop_neg, type = 'b', col = 'red')
legend('topright', legend = c('positive', 'negative'), col = c('black', 'red'), lty = 1)
summary the binary feature by distance
Comment: seems that when the distance is more close, the ATAC feature is more abundant.
summary_by_dist = function(cor_threshold, dist_range){
train_all_sig$atac = ifelse(train_all_sig$correlation > cor_threshold, 1, 0)
prop_pos_by_dist = rep(NA, length(dist_range)-1)
prop_neg_by_dist = rep(NA, length(dist_range)-1)
for (i in 1:(length(dist_range)-1)){
sub_dat = train_all_sig[train_all_sig$tss_dist_to_snp %in% seq(dist_range[i], dist_range[i+1], by = 1), ]
sub_pos = sub_dat[sub_dat$y == 1, ]
sub_neg = sub_dat[sub_dat$y == 0, ]
prop_pos_by_dist[i] = sum(sub_pos$atac == 1)/dim(sub_pos)[1]
prop_neg_by_dist[i] = sum(sub_neg$atac == 1)/dim(sub_neg)[1]
}
return(list(pos = prop_pos_by_dist, neg = prop_neg_by_dist))
}res1 = summary_by_dist(0.5, dist_range = c(0, 1e4, 5e4, 1e5, 2e5, 3e5, 4e5, 5e5, 3e6))
res2 = summary_by_dist(0.8, dist_range = c(0, 1e4, 5e4, 1e5, 2e5, 3e5, 4e5, 5e5, 3e6))
par(mfrow = c(1,2))
plot(res1$pos, type = 'b', ylab = 'proportion', main = 'threshold=0.5')
points(res1$neg, type = 'b', col = 'red')
legend('topright', legend = c('positive', 'negative'), col = c('black', 'red'), lty = 1, cex = 0.6)
plot(res2$pos, type = 'b', ylab = 'proportion', main = 'threshold=0.8')
points(res2$neg, type = 'b', col = 'red')
legend('topright', legend = c('positive', 'negative'), col = c('black', 'red'), lty = 1, cex = 0.6)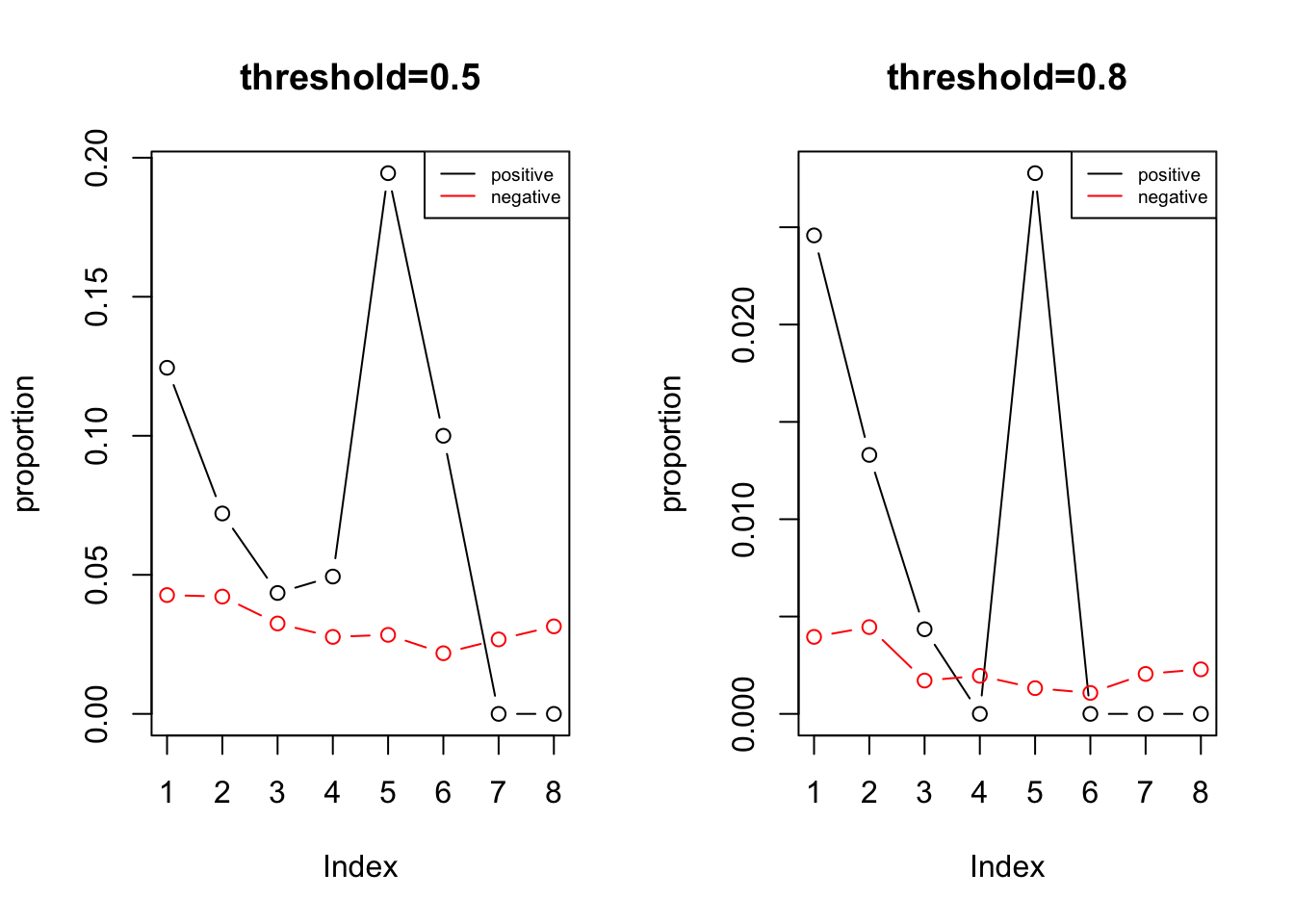
res1 = summary_by_dist(0.5, dist_range = c(0, 1e4, 5e4, 1e5, 3e6))
res2 = summary_by_dist(0.8, dist_range = c(0, 1e4, 5e4, 1e5, 3e6))
par(mfrow = c(1,2))
plot(res1$pos, type = 'b', ylab = 'proportion', ylim = c(0, 0.15), main = 'threshold=0.5')
points(res1$neg, type = 'b', col = 'red')
legend('topright', legend = c('positive', 'negative'), col = c('black', 'red'), lty = 1, cex = 0.6)
plot(res2$pos, type = 'b', ylab = 'proportion', ylim = c(0, 0.03), main = 'threshold=0.8')
points(res2$neg, type = 'b', col = 'red')
legend('topright', legend = c('positive', 'negative'), col = c('black', 'red'), lty = 1, cex = 0.6)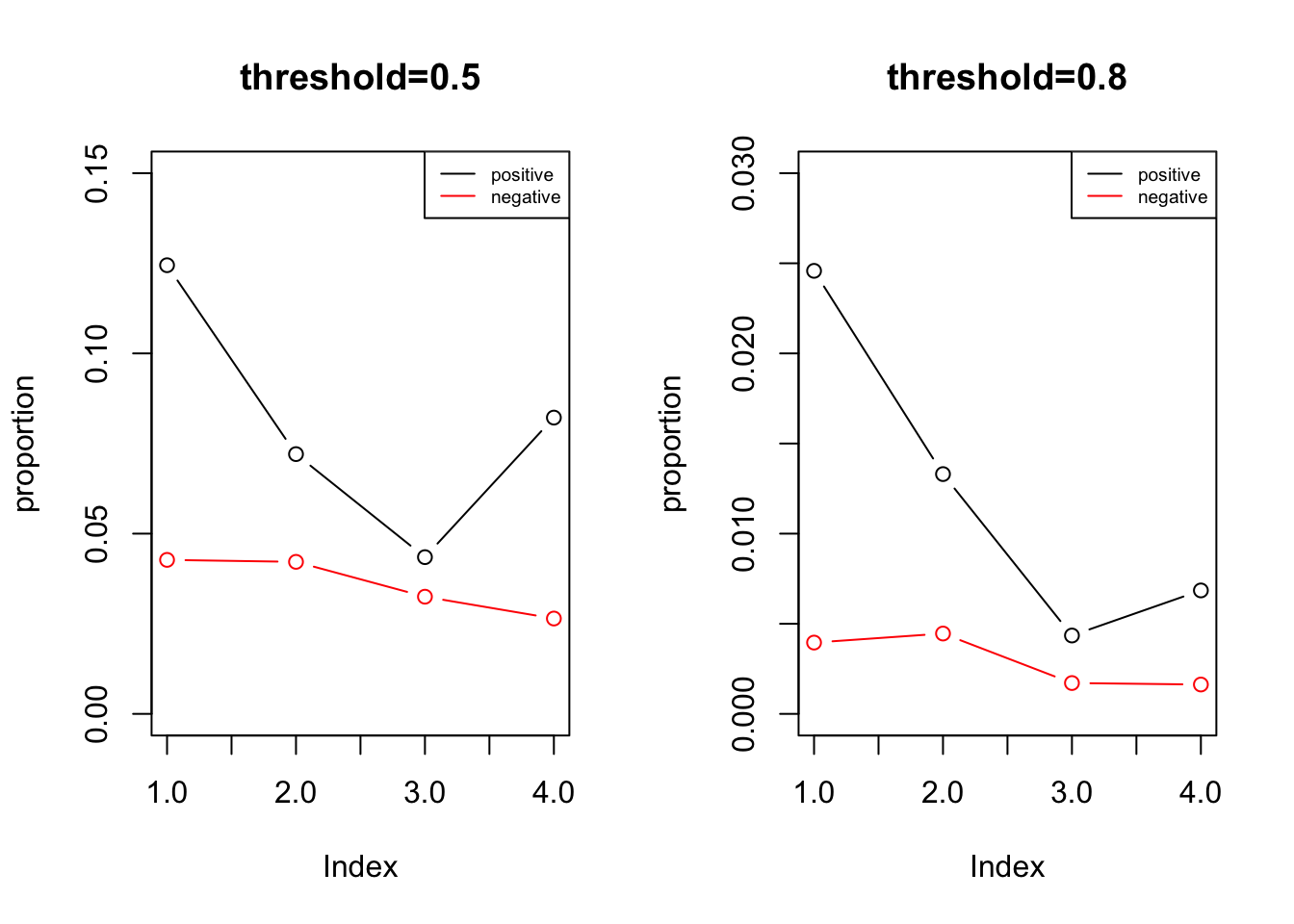
sessionInfo()R version 3.6.3 (2020-02-29)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS Catalina 10.15.5
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] workflowr_1.6.1
loaded via a namespace (and not attached):
[1] Rcpp_1.0.4 rprojroot_1.3-2 digest_0.6.25 later_1.0.0
[5] R6_2.4.1 backports_1.1.5 git2r_0.26.1 magrittr_1.5
[9] evaluate_0.14 highr_0.8 stringi_1.4.6 rlang_0.4.5
[13] fs_1.3.2 promises_1.1.0 whisker_0.4 rmarkdown_2.1
[17] tools_3.6.3 stringr_1.4.0 glue_1.3.2 httpuv_1.5.2
[21] xfun_0.12 yaml_2.2.1 compiler_3.6.3 htmltools_0.4.0
[25] knitr_1.28